|
CYSTIC FIBROSIS: A CASE STUDY IN COURAGE
written
by Kathy
L. Nichols; Graduate Student, University
of Florida
 By
the time her daughter, Katelyn,
was four-and-a-half years old, Leisa
Sims had begun hearing the word "paranoid"
from physicians in response to her concerns
for her daughter’s health (K.
Sims & L. Sims, personal communication,
April 1, 2010). She had been
to doctors a total of 27 times in the
prior 24 months trying to find out what
was wrong, all to no avail. By
the time her daughter, Katelyn,
was four-and-a-half years old, Leisa
Sims had begun hearing the word "paranoid"
from physicians in response to her concerns
for her daughter’s health (K.
Sims & L. Sims, personal communication,
April 1, 2010). She had been
to doctors a total of 27 times in the
prior 24 months trying to find out what
was wrong, all to no avail.
This paper
documents the experience of Katelyn
and her family - starting from those
early days of seeking answers and the
difficulties encountered; moving through
Katelyn’s school years, including
some incredibly positive experiences,
as well as some incredibly painful ones;
and concluding at the present, with
Katelyn now 19 ½ years old and
living an active life as a college student,
employee, and spokesperson for the Cystic
Fibrosis Foundation. Woven throughout
are clinical details about cystic fibrosis,
including the importance of exercise
therapy and physical activity in treating
the disease.
DIAGNOSIS
Though Leisa
Sims had tried for two years to find
out why Katelyn - who weighed a scant
23 pounds at four-and-a-half years old,
and who was almost constantly on antibiotics
- was sick and frail, it was not until
an advanced registered nurse practitioner
(ARNP) closely examined Katelyn, noting
specific signs such as clubbing of her
digits, a protruding belly, and a deep
cavity in her ribcage, that suspicions
of cystic fibrosis (CF) were raised.
The early signs and symptoms of CF vary
and display at different times from
person to person (National
Heart, Lung, and Blood Institute (NHLBI)
Signs and Symptoms, 2009; Mayo
Clinic, 2010). In addition to
the signs the ARNP noted in Katelyn,
other signs of CF can include a salty
taste to the skin, repeated lung and
sinus infections, and a lack of stools
for newborns or very frequent loose
and bad-smelling stools for slightly
older children (NHLBI
Signs and Symptoms, 2009; Mayo
Clinic, 2010).
According
to the Cystic Fibrosis Foundation (CFF),
CF is an inherited chronic disease that
affects the respiratory and digestive
systems of approximately 30,000 adults
and children in the United States, with
1,000 new cases being diagnosed each
year (2010). A defective gene and its
protein product cause the body to produce
unusually thick, sticky mucus that:
- clogs the lungs and leads to life-threatening
lung infections; and
- obstructs the pancreas and stops
natural enzymes from helping the body
break down and absorb food. (CFF,
2010)
The gene in
question is the CFTR (cystic
fibrosis transmembrane regulator)
gene, which resides on the seventh pair
of chromosomes (University
of Virginia Health System, 2008).
Errors or mutations in this gene cause
CF, over 1,000 types of which already
have been identified. The University
of Virginia Health System (UVaHS) notes
that CF is an "autosomal
recessive" condition (2008).
"Autosomal" means that the
gene is found within the first 22 chromosomes,
which have no determination for gender,
and therefore affects males and females
equally. "Recessive" means
that two copies of the gene, one from
the mother and one from the father,
are prerequisite to having the condition
(UVaHS, 2008).
If a person
receives only one copy of the mutated
gene, he or she will be a "carrier"
of the disease, but will be healthy
and not likely to know of the carrier
status (UVaHS,
2008). As with other autosomal
recessive conditions, this is why in
eight out of ten cases, there is no
family history of CF and the diagnosis
is completely unexpected (as was the
case with the Sims family). Once parents
have had a child with CF, the likelihood
of a CF diagnosis with each future pregnancy
is 25 percent, or in other words, a
three out of four chance that another
child will not have CF. One's ethnic
background, however, contributes to
whether one carries a mutated gene,
and, therefore, affects the risk of
having a child with CF
(UVaHS, 2008). Those of Caucasian
and Ashkenazi Jewish background have
a significantly higher likelihood of
carrying a mutated gene, as shown in
Table 1 below.
Table
1
Risk of Having
a Mutation in the Gene for CF and of
Having a Child with CF by Ethnic Background
(with no family history of CF) (UVaHS,
2008)

CF is sometimes
misdiagnosed as asthma, as both diseases
block the airway and can cause wheezing,
coughing, and dyspnea (Weinberger
& Abu-Hasan, 2007). When
CF is suspected, however, it can typically
be confirmed by a "sweat test"
that indicates if the levels of sodium
and chloride in one's sweat are higher
than normal, a condition indicative
of CF (UVaHS,
2008). To ensure a valid and
reliable measurement is made, avoiding
false-positive and false-negative results,
duplicate collections are required to
be taken and analyzed (Weinberger
& Abu-Hasan, 2007).
Presuming substantial
agreement between the results for the
samples measured, a sweat chloride concentration
of 60 mEq/L generally indicates cystic
fibrosis. Concentrations of <40 mEq/L
generally reassure that cystic fibrosis
is not the cause of the symptoms. Levels
of 40 to 60 mEq/L (>30 mEq/L for
infants) are sufficiently suspicious
to warrant genetic analysis to determine
whether there are two mutations of the
CFTR gene (Weinberger
& Abu-Hasan, 2007). Other
diagnostic procedures that can be used
include chest x-rays, lung function
tests, chemical tests, sputum cultures,
and stool evaluations (UVaHS,
2008).
While the lungs
and pancreas are the most frequently
affected organs (CFF,
2010), as shown in Section A
of Figure 1, other organs may also be
involved. Sections B and C of the figure
demonstrate the difference between a
normal airway and an airway with cystic
fibrosis, including the effects of mucus
build-up leading to bacterial infection.

Figure
1. Organs Affected by Cystic Fibrosis
and Airway Obstruction Leading to Bacterial
Infection (NHLBI
Signs and Symptoms, 2009)
TREATMENT
Once a diagnosis
of CF has been made, treatment to offset
the effects of the disease begins. For
the families affected, not only do they
have to deal with the emotional impact
of the diagnosis, they also have to
learn how to provide a strict treatment
regimen for the family member with CF
and implement that regimen right away.
While each person’s treatment
plan is individualized based on needs
and limitations, the overall goals of
CF treatment are to prevent and manage
lung infections, intestinal blockages,
and dehydration; loosen and eliminate
thick mucus from the lungs; and provide
sufficient nutrition (NHLBI
CF Treated, 2009). There are
a variety of ways that these goals can
be met.
In kindergarten,
Katelyn
Sims’ daily treatment regimen
included several components: nebulizer
treatments, digestive enzymes, chest
physiotherapy (CPT), and general physical
activity. These were no small feats
to accomplish considering that some
of the enzymes and nebulizer treatments
had to be administered during school
hours, without the school’s assistance.
This meant that Leisa Sims had to make
three trips to the school each day in
addition to the trips for morning drop-off
and afternoon pick-up.
This was not
counting the multiple CPT sessions Katelyn
needed in and around school time, which
were where Leisa would administer percussion
and postural drainage, or pounding Katelyn’s
chest and back repeatedly with cupped
hands to loosen the mucus from her lungs
so it could be expectorated (NHLBI
CF Treated, 2009). It also didn’t
account for the extracurricular physical
activities that Katelyn engaged in,
such as competitive pom-pom dancing
competitions, at which she excelled
despite CF. For instance, Katelyn and
her pom-pom team won a state competition
not long after she was diagnosed, even
though Katelyn had a peripherally inserted
central catheter (PICC) line in place
for the administration of antibiotic
at the time.
Even the components
of Katelyn’s treatment plan that
sounded fairly innocuous, such as taking
digestive enzymes, were difficult. At
four years old, Katelyn had a prescription
for 1,500 enzyme capsules per month,
but because she was so active, and because
her body didn’t properly process
the nutrients in the foods she ate,
she was (and still is) constantly hungry,
and almost always exceeded the limits
of the prescription. This meant Katelyn
was ingesting more than 50 enzyme capsules
per day. At first her mother would mix
the contents of the capsules into foods
like apple sauce because Katelyn could
not swallow the capsules whole, but
by six years old, Katelyn was so experienced
at taking the capsules that she could
ingest 10 at a time independently.
While Katelyn’s
neuromuscular, organic, and interpretive
physical education objectives were being
met, during her kindergarten year she
had a crisis of sorts when it came to
the social and emotional aspects of
the therapies she was beginning to understand
were essential for her to survive. Leisa
Sims had originally encouraged Katelyn
to be home schooled but acquiesced when
Katelyn - who has a very close bond
with her older sister, Amber - asked
to be able to go to school with other
kids just as Amber had done.
When Katelyn’s
teacher, who had noticed the toll the
constant school visits by Leisa Sims
were taking, offered to administer the
nebulizer treatments and enzymes during
school hours but was told by the school
administration that they would not allow
it, Katelyn concluded that the school
was not in support of her being able
to survive, and she asked to be removed
from the school. She did not return
to a traditional school setting again
until fourth grade, though it was to
a different school.
In the meanwhile,
as Katelyn was homeschooled, she remained
physically active but was declining
in health. At eight years old, however,
Katelyn had the opportunity to participate
in a clinical trial involving hormone
therapy. From an early traumatizing
experience with IVs in a children’s
hospital, Katelyn had more than the
usual share of fear when it came to
needles, and typically required valium
to get through shots and IV lines being
placed. The clinical trial involved
hospitalization for a two-day period
four times per year, during which complete
fasting was required from 6:00 a.m.
to 2:00 p.m. (a major difficulty for
CF patients), and blood was drawn every
10 minutes.
During the time
not spent in the hospital, three months
of daily shots were required, and then
three months of ingesting a powder.
Leisa Sims let Katelyn know that the
decision to participate was hers to
make, and that there was no pressure
for her to decide one way or the other.
After some thought, and with her usual
resilience and courage, Katelyn informed
her mother that even if it didn’t
help her, it might help others, so she
was going to do it.
The course of
the trial was incredibly difficult for
Katelyn, but in the long run, it was
well worth it. Near the start of the
trial, Katelyn’s parents had finally
found out a prognosis for their daughter,
which they had until that time decided
not to pursue. They were told that Katelyn
was not likely to make it past 12 years
old based on her condition at that time.
She was in the 25th percentile for weight,
short in stature, weak, and frail. Over
the course of the trial, though, Katelyn
grew taller, her shoe size went from
a two to a ladies size seven, and she
gained 13 pounds, putting her in the
75th percentile for weight. Perhaps
most remarkable, though disheartening
at the same time, Katelyn was the only
one out of the 20 children who participated
in the clinical trial for whom the treatment
worked.
The clinical
trial was a turning point for Katelyn,
and with renewed vigor, she was able
to enjoy the sports and activities she
liked, such as gymnastics, dance, baton
twirling, basketball, and swimming.
During her hospital stays, which occurred
approximately once every 14 months at
that point, Katelyn began writing poetry,
which helped her to express her feelings
and work through some difficult times.
One of those times occurred in fourth
grade when a classmate told Katelyn
that she was going to die soon, and
then, after Katelyn’s dad, David,
and Leisa Sims had helped Katelyn through
that hurtful instance, the same student
told her that he would be glad when
she died. Despite these types of challenges,
though, Katelyn stuck it out and stayed
in school.
Not only did
Katelyn stay in school, she accomplished
more than the majority of her peers
despite having to contend with CF on
a daily basis. As a testament to her
success, Katelyn has over 250 trophies
from the various activities in which
she participated. In high school, for
instance, Katelyn was consistently on
the honor roll, parliamentarian in Future
Farmers of America all four years, voted
best poet in the school, a member of
the prom and homecoming courts, captain
of the varsity cheerleading squad and
dance team, winner of the Bradford Union
County Strawberry Pageant, and much
more. She even managed to squeeze in
work as a hostess at a local barbecue
restaurant and as a runway model for
bridal shows. Despite missing 64 days
her senior year due to medical absences,
Katelyn made straight A’s that
year and graduated with a 3.4 grade
point average.
Even as Katelyn
participated in all of these activities,
she was still undergoing daily treatments
for CF, including chest physiotherapy,
breathing exercises, and enzyme supplementation.
For the chest physiotherapy and breathing
exercises, the time frame involved can
be roughly an hour to an hour and a
half daily. These types of Airway Clearance
Therapies (ACTs) can be accomplished
via a variety of methods, none of which
is better than the other in terms of
efficacy (Lester
& Flume, 2009). There are
significant differences, however, when
it comes to the appropriate age for
use and the advantages and disadvantages
of the various types of ACTs as shown
in Table 2.
Table
2
Airway Clearance Methods (Lester
& Flume, 2009, p. 735)
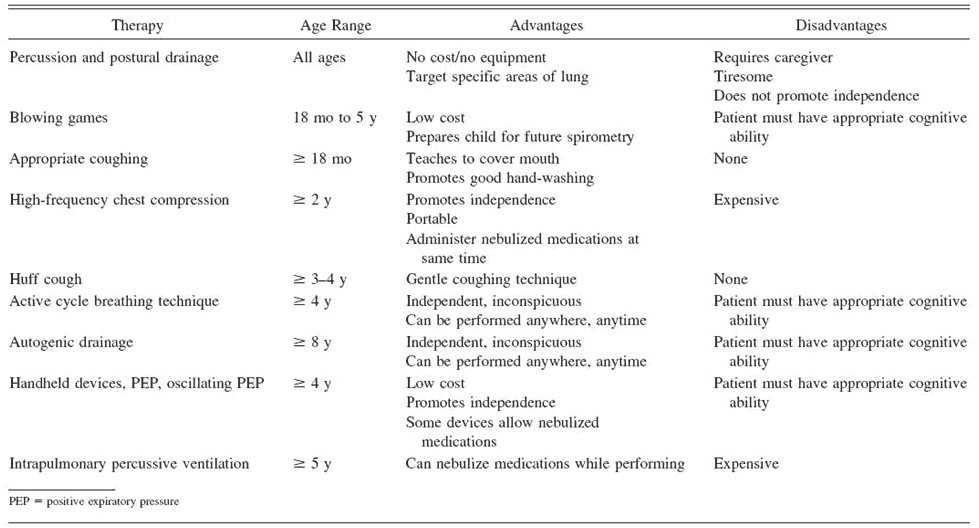
Clearing airways
is a critical part of treatment for
CF. Patients learn a variety of breathing
and coughing techniques to help loosen
and remove secretions. Various types
of equipment are also available to help
in the process. For instance, mechanized
vests provide high-frequency chest compression
taking the place of manual percussion
and postural drainage sessions (Lester
& Flume, 2009). The vests
inflate and deflate to various frequencies
(Hz), pressing and releasing the chest
wall, which in turn helps to dislodge
secretions that can then be coughed
out. One disadvantage is the expense
of the vests. In Katelyn’s case,
her family did fund-raising to be able
to buy her a vest when she was ten years
old. Though she will sometimes use the
vest in the mornings, the weight and
bulkiness of the vest are deterrents
to her for its use, and she prefers
manual percussion and postural drainage
when possible.
Another type
of ACT is Positive Expiratory Pressure
(PEP) (Lester
& Flume, 2009). Hand-held
devices are available that provide resistance
when they are blown into, causing back
pressure in the lungs and getting air
behind secretions. Some of these devices,
including the one preferred by Katelyn,
also have oscillating internal parts
that help to loosen phlegm by vibrating
the airway walls upon exhalation.
Intrapulmonary
percussive ventilation (IPV) is an ACT
method used primarily in hospitals for
patients who have increased secretions
that they are unable to clear on their
own (Lester &
Flume, 2009). It involves a pneumatic
device that delivers 100 to 300 short,
high-flow air bursts to the lungs a
minute. At the same time, an aerosol
mist is delivered to loosen secretions.
An addition
to ACTs, two other critical components
of care are to avoid or treat both malnourishment
and lung infections as rapidly and effectively
as possible (Steinkamp
& Wiedemann, 2002). Steinkamp
and Wiedemann found that over a two-year
period, and for all age groups, patients
of normal weight experienced significantly
less decrease in lung function than
those with malnutrition (2002). Leisa
Sims recounts Katelyn’s eating
habits with a smile, as she notes that
Katelyn can easily eat two foot-long
subs, potato chips, and a large soda,
and then 30 minutes later eat a bowl
of cereal and still be hungry for more.
In terms of
lung infections, of particular concern
to those with CF is the opportunistic
infection Pseudomonas
aeruginosa. According to Todar’s
Online Textbook of Bacteriology,
Pseudomonas aeruginosa "...exploits
some break in the host defenses to initiate
an infection. In fact, Pseudomonas aeruginosa
is the epitome of an opportunistic pathogen
of humans. The bacterium almost never
infects uncompromised tissues, yet there
is hardly any tissue that it cannot
infect if the tissue defenses are compromised
in some manner" (Todar,
2008, p. 1). Steinkamp and Wiedemann's
conclusion: the absence of Pseudomonas
aeruginosa infection and the presence
of normal body weight were indicative
of better preservation of lung function
(2002).
At times, managing
ACTs, nutrition, and infection means
a hospital stay for people with CF.
For instance, from age eight to 11,
Katelyn had a hospital stay approximately
once every 14 months. When she was 11
to 13 years old, the visits averaged
one to two times per year. From 13 years
old to the present, the visits have
been every one to three months. These
hospital stays typically last a minimum
of two weeks each. In addition to these
hospital stays, Katelyn makes a monthly
hospital check-up visit, which includes
having her portacath flushed and engaging
in consultations with nutritionists,
pulmonologists, endocrinologists, gastroenterologists,
and behavioral pediatricians. With all
of the hospital stays and check-up visits,
it is even more impressive to note what
Katelyn has managed to accomplish with
the last piece of the treatment puzzle
for CF: Physical Activity.
In addition
to participating in dance, gymnastics,
baton twirling, cheerleading, basketball,
and swimming, Katelyn has also played
softball and done weight lifting. While
the Cystic Fibrosis Foundation recommends
that people with CF first check with
their doctors to ensure they are in
sufficient condition before engaging
in formal exercise programs (Cerny,
2009), the organization also
clearly conveys the benefits of exercise
to those with CF:
Children,
teens and adults with CF who exercise
do better than those who don’t.
Their rate of lung function decline
slows. They enjoy a more normal lifestyle.
Regular exercise helps the heart so
it is stronger during stress. Regular
exercise also helps the lung function
so there are more reserves to use
during exacerbations, or lung infections.
(Cerny, 2009,
p. 1)
This recommendation
matches that of the Clinical Practice
Guidelines for Pulmonary Therapies Committee,
which concluded that aerobic exercise
should be used as an adjunctive therapy
both to help clear airways and for overall
health benefits (Flume
et al., 2009). The Cystic Fibrosis
Foundation does note, however, the particular
importance of building up stamina and
watching out for heat stress in terms
of the extra sodium and chloride lost
in CF sweat (Cerny,
2009).
FUTURE
DIRECTIONS
Some promising,
yet in some ways controversial, treatments
currently are being examined for CF
patients. One of these is lung transplantation
(Adler et al.,
2009). Although some CF patients
have already received transplanted lungs,
issues such as organ allocation and
potentially poor survival benefit make
this a somewhat contested treatment
option (Adler
et al, 2009).
One of the more
interesting and innovative treatments
currently being researched is a gene
therapy approach that converts the thick,
sticky mucus of CF patients (that traps
pathogens and fosters infection) into
the thin, slippery mucus of healthy
people (that moves pathogens out of
the body via cilia) (Meadows,
2009). Though not yet tested
on humans, researchers have been able
to do this in the lab in the short term
by inserting properly functioning CTFR
into human parainfluenza virus (PIV)
- a cause of the common cold and respiratory
infections - and using the virus as
a delivery mechanism to get the CTFR
to cultured cells that simulate the
airway lining (Meadows,
2009). The results of this research
look promising, though a lot of fine
tuning will be needed before the medical
community will embrace this treatment.
So what is next
for Katelyn Sims? She is taking classes
at Santa Fe College and has ambitions
to be a marine biologist and research
ocean populations if she is able to
get SCUBA certified. If that is not
possible, she has a back-up plan, which
is to study ballistics. She still works
as a hostess for a restaurant and does
modeling for bridal shows. She’s
also a formidable spokesperson for the
Cystic Fibrosis Foundation, having raised
many thousands of dollars on behalf
of the organization. Last year, for
instance, Katelyn spearheaded the inaugural
Bradford
County Great Strides Walk for a Cure,
in which 258 people participated, raising
$15,300 for research to find a cure
for CF. This will be an annual event
from now on, with plans already set
for this year’s walk, scheduled
for September 11, 2010.
This extraordinary
young woman, whose understated demeanor
belies her incredible spirit and courageous
zest for life, sums it all up with her
philosophy: Life might not be the
party we hoped for, but while we’re
here, we might as well dance.
And Katelyn
is one remarkable dancer.
references
|

